An insightful presentation that explains how FDA reviewers examine a submission.It includes 3 practical ideas for how to create a better submission. These ideas apply to submissions for drugs, biologics, and medical devices. Over 1,000 companies have paid to hear this presentation.
Joshua Sharlin’s Credentials
Summary of Credentials:
- Worked both sides of the regulatory fence and has both prepared
and reviewed submissions - Former FDA reviewer responsible for examining statistical and safety and efficacy data
- Former instructor for SAS Institute with over 20 years of SAS programming experience
- Has helped numerous pharmaceutical and medical device companies develop and improve the content of their submissions
- Has taught thousands of professionals in FDA-regulated firms covering many areas of regulatory affairs, Part 11 assessment and compliance, software validation, improving the quality and efficiency of software development, and developing, evaluating and improving company SOPs
- Before joining FDA, was Director of Hotel Customer Information Systems
at Marriott Hotels
Intended Audience
Everyone interested in developing submissions responsive to the needs of FDA reviewers and therefore reducing the time to product approval will benefit from this presentation.
- Staff and managers responsible for:
- Regulatory affairs
- QA/QC
- Information systems/information technology
- Clinical trial data
- GMPs (Good Manufacturing Practices)
- Compliance
- Software developers
- Medical writers and editors
Objectives of This Presentation
- Present general principles outlining how to be more effective
with FDA - Present ideas applicable to all types of submissions for drugs, biologics, and devices
- Improve understanding of how FDA reviewers do their job
- Learn how to add information to a submission to help reviewers work more quickly
- Examine how to be more efficient in preparing a submission
- Study practical examples of information content and ideas for organization you can apply to your own data and situations
- Emphasize the difference between finishing a submission and obtaining FDA approval
- NOT to give specific advice on a specific type of submission
- NOT to review FDA regulations
Requirements, Standards and Guidances for Preparing a Submission (Diagram)
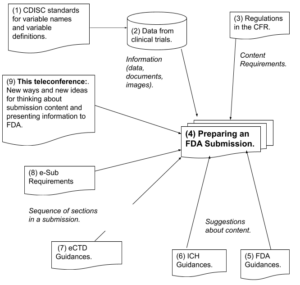

CDISC – Clinical Data Interchange Standards Consortium
CFR – Code of Federal Regulations
CTD – Common Technical Document
ICH – International Conference on Harmonization (USA, EU, Japan)
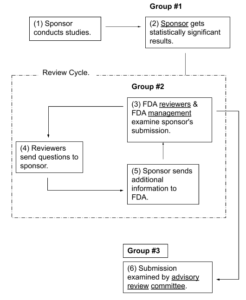
Three Groups Examine Submission Information

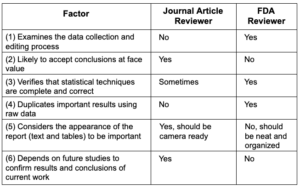
Writing a Journal Article is NOT like Preparing
an FDA Submission
Two reasons most submissions fail to be “reviewer-friendly”
- Work is performed too quickly during the last few months
of submission preparation - Sponsor does not acknowledge the difference between a manuscript
for publication and a submission to FDA
Sponsors sometimes consider an FDA submission to be the same as a very large manuscript submitted to a refereed scientific journal. In fact, the two
are actually quite different.

Companies and FDA Are Mutually Dependent
on Being Successful
- Companies are dependent on FDA reviewers for product approval
- FDA reviewers are dependent on the quality of company submissions to meet performance goals
After Information Collection is Completed,
There are Two Opportunities to Reduce Approval Time
- Increase the efficiency of producing the submission
- Reduce the time needed by FDA reviewers to examine a submission by eliminating review cycles
Reducing the Time Needed by FDA Reviewers
to Examine a Submission: The Overlooked Opportunity
- The overlooked opportunity
- Many companies assume that the FDA review process
is unknowable and can’t be speeded up - Can become a reasonable goal if one understands how
reviewers think and work
The Strategy for Reducing FDA Approval Time
- Understand how FDA reviewers think
- Examine how FDA reviewers work
- Apply these concepts to reducing the time needed to produce a submission and the time used by FDA to review the submission
Four Factors Influencing How Reviewers Think
Reviewers are concerned with lots of areas and many are very subject specific, but these areas can be generalized into four topics:
- Safety: Is the drug/device/biologic safe?
- Efficacy: Does the drug/device/biologic do what the label says?
- Accountability: Can I hold someone responsible for key decisions and actions?
- Trust: Are the company’s results and conclusions believable?
How Reviewers Work
What reviewers do when they turn on the lights in the morning?
They investigate:
(1) Process Execution
(2) Process Results
(3) Process Problems
How Reviewers Work: (1) Process Execution
The Reviewer Issue:
Examine process execution.
Compare what was supposed to be done to what was actually done.
– Are they the same?
The Reviewer Challenge:
Determine that the process was executed correctly.
How Reviewers Work: (2) Process Results
The Reviewer Issue:
Reviewers selectively duplicate process results for two reasons:
- To gain confidence in the accuracy of information
- To get a complete understanding of how the work was done
The Reviewer Objective:
Manipulate information for insight into the subtleties and complexities of submission contents.
How Reviewers Work: (3) Process Problems
The Reviewer Issue:
Learn about problems and issues (known, potential, and unresolved) associated with the process being examined.
The Reviewer Challenge:
- Become confident all problems have been identified
- Be sure problems and issues are answered and solved correctly
- Communicate review status to FDA upper management
Following Only Guidance Documents
& the Code of Federal Regulations (CFR) is Insufficient
Problem Statement
- Many submissions have significant problems with quality
- The needs and the complexities of reviewer’s work
is not addressed in guidance documents and the CFR
Solution
- Companies need to be creative in tailoring information presentation and organization to show their data in the
best possible way. - The submission should allow reviewers to quickly:
- Find the information they need to understand process execution, process results and process problems
- Agree with the conclusion that the product is safe
and effective
Techniques Apply Equally to FDA Reviewers
and Drug/Biologics/Device Sponsors
The following examples illustrate ideas for better efficiency
and improved processes and apply equally to:
- Drug, device and biologics sponsors building a submission
- FDA reviewers examining a submission
Helping Reviewers Understand Process Execution: Example #1 – Create High Level Flow Chart
Problem
Reviewers need to:
- Quickly understand the overall structure and organization of a trial
- Communicate basic information about the trial to FDA management
Solution
- Sponsors should prepare a high level flow chart
- A series of hierarchical flow charts may be necessary
High Level Flow Chart, Example
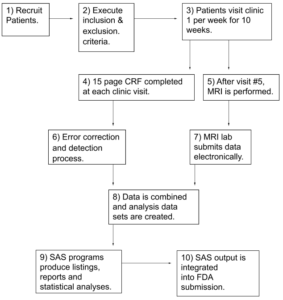

CRF- Case Report Form. A paper or electronic form for recording information about a patient
MRI – Magnetic Resonance Imaging
SAS- Software used by almost all companies to conduct data manipulation and prepare tables, listing and statistical analyses for a submission
Helping Reviewers Understand Process Execution: Example #2 – Organize and Include Correspondence
Problem
Reviewers need to:
- Read past correspondence to confirm facts or sequences of events. For example:
- Notes from meetings and phone calls between FDA
and the sponsor - Letters discussing the submission
- Find specific correspondence quickly
- Within FDA, correspondence can be filed and organized poorly
Solution
- Add an appendix to your submission including all correspondence organized in chronological order
- Use a descriptive Table of Contents which includes the “to” and “from” and a brief summary of what the correspondence covers
Helping Reviewers Understand Process Results:
Example #1 – Create Summary Information about Data
Problems – Reviewers need to:
- Learn about details of data that contain study results. Understanding basic information can be time consuming
- Answer questions from other reviewers and FDA management about how data was handled and processed and how information management problems were solved
Solutions – Sponsors can:
- Create tables and listings of summary information about key sets of data (could be SAS, Oracle or anything else)
- For example, produce a Data Management Appendix that contains for each dataset:
- List of variables and their meaning or calculation. This is in the guidance document for electronic submission content
- Identification of the variables which create a unique identifier for each observation
- Frequency distributions for discrete variables.
- Descriptive statistics for continuous variables
- Print of the first 25 rows or observations
Oracle – Database product used by many companies to store submission information
SAS – Software used by almost all companies to conduct data manipulation and prepare tables, listing and statistical analyses for a submission
Helping Reviewers Understand Process Results:
Example #2 – Flowchart Showing How Critical Output
Was Produced
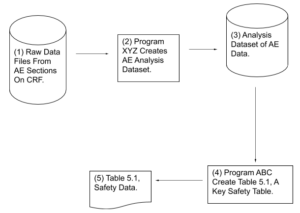

CRF – Case Report Form. A paper or electronic form for recording information about a patient
AE – Adverse Event
Helping Reviewers Understand Process Results:
Example #3 – Confirm Programs Produce Correct Output
Problem – Reviewers need to:
- Reproduce key safety and efficacy results using the SAS datasets and programs supplied by the drug/device sponsor
- Avoid time consuming problems understanding why they can’t reproduce the sponsor’s results using the sponsor ‘s programs and data
Solution – Sponsors can:
- Recreate the reviewer’s computing environment (i.e., stand-alone desktop)
- Re-run all programs
- Confirm that the recreated output matches information in the submission
- Perform this work before the submission is sent to FDA
SAS – Software used by almost all companies to conduct data manipulation and prepare tables, listing and statistical analyses for a submission
Helping Reviewers Understand Process Problems: Example #1 – Track Known Problems
Method: Create a Known Problem Spreadsheet
Convert knowledge about known problems into an advantage.
Introduction:
- These problems are often discovered after submission preparation is well underway
- Many companies don’t mention these problems, hoping the issue will not be discovered by the reviewer
Solution:
- Create a known problems resolution spreadsheet. Steps:
- Identify known problems with data or execution of the trial
- Explain what happened and describe the impact on results, especially safety and efficacy
Conclusion:
Don’t force the reviewer to spend time discovering and understanding study problems.
Known Problems Spreadsheet – Example
| (A) Known Problem | (B) Effect on Conclusions About Efficacy | (C) Effect on Conclusions About Safety | |
| (1) Patient 101 took medications out of sequence | None, the action was within limits set by the protocol | ||
| (2) Patient 33 was started on the study even though they did not meet inclusion criteria | The patient did meet all inclusion criteria within the first two weeks of the study start. | ||
| (3) SOP defining visit windows was not followed for the first month of the study | The SOP could not be followed because the medical isolation room was under construction. Adjustments were made that were within protocol specifications. | ||
| (4) Patient 410 took a prohibited concomitant medication during week 5 | There was an adverse event during week 5 which was attributed to the prohibited concomitant medication |
Helping Reviewers Understand Process Problems: Example #2 – Track Potential Problems
Method: Create a Potential Problem Spreadsheet
Introduction:
- Identify issues that could be interpreted as a problem
- Explain why it wasn’t a problem
Solution:
- Be specific about why there was no effect on either safety or efficacy
Conclusion:
It’s frustrating for a reviewer to spend time investigating a “problem” only to discover that with more knowledge it is apparent there was no problem after all.
Potential Problem Spreadsheet, Example
| (A) Potential Problem | (B) Why the problem had no effect on either safety or efficacy | (C) Effect on Conclusions About Safety or Efficacy |
| (1) Patients in the treatment group had twice as many adverse events as the control group | The treatment group was followed for three years longer than the control group, so it expected that they would have more adverse events | None |
| (2) Patient 45 missed ten clinic visits but was not dropped from the study | Patient 45 suffered a broken leg and could not travel to the clinic. However, the same treatments were given at home. | None |
| (3) Patients 90 and 95 were given two doses of drug during the third visit. | For these two patients, the drug vials were dropped and broken and could not be given to the patient. To maintain proper drug inventory records these quantities of drug were assigned to these patients. | None |
Helping Reviewers Understand Process Problems: Example #3 – Track Unresolved Problems
Example: Create a Problem Resolution Spreadsheet
Tracking Problems During the Execution of a Process and the Production of Information for FDA:
- Part of the flow of information between the sponsor and FDA can be viewed as a series of problems to be understood and solved.
- Reviewers must identify all problems that represent barriers to interpreting information.
- Sponsors must understand the problems and provide data and information for their resolution.
- For this process to work efficiently, both groups should agree on the description and the status of all problems.
- Changing staff and a review process that can take years adds to the challenge of managing this information.
- A spreadsheet format is an ideal way to create an inventory of problems and track their evolution and resolution.
Problem Resolution Spreadsheet
(Tracking unresolved problems)
| (A) Pre-IND Meeting Held at FDA on January 20, 2017 | (B) June 3, 2017 Letter to Company From FDA | (C) August 10, 2017 Meeting at FDA To Review Outstanding Problems | |
| (1) Give reasons for deleting patients 101 and 220 from efficacy analysis | Medical officer asked for more info about patients 101 and 220 | Copies of CRFs were provided but FDA wants more information | |
| (2) Provide more information about how patients were randomized to treatments | Problem first mentioned by FDA | Randomization procedure was described and FDA had no further comments | |
| (3) Explain why drug stability study ended after 6 months | CMC reviewer asked for more information | FDA accepted reasons for 6 month study |
IND – Investigational New Drug. A type of submission required before a study using people can begin.
CRF – Case Report Form. A paper or electronic form used to record information about a patient.
CMC – Chemistry, Manufacturing and Controls. A subject area within a submission examined by FDA reviewers.
Overlooked Opportunities to Improve Your Submission and Interactions with FDA Reviewers
- Be aware that rushing your submission creation efforts near your artificial submission deadline creates mistakes. Solution: Always work slowly and carefully.
- Recognize what reviewers really mean when they use the word “suggest.” (it means “do this”)
- Understand the benefits of submitting SAS code that created the analysis dataset containing the primary efficacy variable.
- When responding to a reviewer’s request for information, ask for a problem statement.
- Questions to reviewers should be multiple choice and not fill-in the blank.
Convincing an FDA Reviewer
- Present overwhelming evidence to prove your point:
- Cite scientific publications
- Reference established facts
- Use FDA precedents
- Present both sides
- Create a powerful enough argument so that anyone who disagrees appears unreasonable.
Summary: What Needs to be in a Product Submission
How Reviewers Work
- Process Execution
- Process Results
- Process Problems
Process Execution
- Create a high level flow chart
- Organize and include correspondence
Process Results
- Create summary information about data
- Flowchart how critical output was produced
- Confirm programs produce correct output
Process Problems
- Track known problems
- Track potential problems
- Track unresolved problems
Transport File – An electronic file format that FDA now requires when sending data to the agency as part of an electronic submission. Will eventually be replaced by a requirement for files in a XML format.
Conclusions
-
- Companies and FDA are mutually dependent for success
- Think like reviewers think and read what reviewers read
- Opportunities to improve submissions have been well-defined
- Meet with FDA early and often and establish written agreements
- Thoroughly understand your regulatory requirements and responsibilities
(and FDA’s) - Respond to every FDA concern in detail
- Think of FDA reviewers as partners, not adversaries
- The examples presented here are intended to be representative and not all-inclusive
- Look for lots of small, submission-specific opportunities for improvement
- It is possible to produce a better submission AND spend less time and money
Next Steps
- For Management:
Consider the possibility of having product approval as a goal rather than simply sending a submission to FDA.
- For Staff:
Increase the quantity and quality of your interactions
with FDA reviewers.
- For Everyone:
Thoroughly learn the rules and submission content requirements for both FDA and your own firm.
Joshua Sharlin, Ph.D., CV
Washington, D.C. ♦ C: 410-231-8900 ♦ jsharlin@pipeline.com ♦ www.SharlinConsulting.com
(page 1 of 3)
FDA Regulatory Expert Witness Summary
Provide Food and Drug Administration (FDA)-related regulatory support to attorneys in cases involving; (i) death or injury caused by drugs, biologics or medical devices, (ii) patents, (iii) insurance claims, (iv) wrongful termination, (v) trade secrets, (vi) merger and acquisitions. Expert in data integrity and software development. Talented in testimony and depositions.
Hands-on experience in the entire lifecycle of FDA-regulated product development, from creating an initial regulatory strategy, thru data collection and analysis, to review and approval at FDA.
Skilled at writing expert reports that describe and explain regulatory and compliance-related issues involving FDA-regulated companies. Specialist in analyzing FDA compliance information to answer three questions: 1) What did the company know and when did they know it? 2) What should the company have known and when should they have known it? 3) What should the company have done and when should they have done it? (See page 3 for a description of expert witness projects.)
Key Skills, Experience & Qualifications
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Work History
Consultant & Principal, 06/1994 to Current
Sharlin Consulting – Washington, D.C.
- In August 2019 completed a 3-year contract as a Drug Development Leadership Advisor. Advise senior Department of Defense leadership on tactical and strategic regulatory actions affecting the path to FDA approval of 19 products (drugs, vaccines and medical devices) under development as medical countermeasures. Write white papers for all programs identifying regulatory strengths, weaknesses and risks affecting progress toward FDA approval. Create and implement recommendations for improvement.
- Expert witness in court regarding matters related to FDA’s regulatory process for drugs, biologics, and medical devices.
- Audit drug companies, contract research organizations (CROs), clinical sites, labs and software vendors for good clinical practice (GCP) compliance. Identify and close compliance gaps in anticipation of an audit by FDA.
- Write, review and improve INDs, NDAs, IDEs, PMAs, 510(k)s, SAPs, protocols and SOPs.
- Solve compliance problems identified by FDA auditors and answer questions posed by FDA reviewers.
- Develop and present more than 40 FDA-related technical, regulatory and compliance topics to over 50,000 people.
- Assist drug companies and CROs in FDA compliance of electronic records, software products, databases and software development.
- Investigate, analyze, and improve data quality and data integrity in FDA-related activities.
- Prepare information for, and present information at FDA meetings with drug, biologic, and medical device companies
Drug Reviewer, 05/1992 to 06/1994
Food and Drug Administration (FDA) – Rockville, MD
Manage the drug review process. Instruct firms on how to proceed with drug approval. Summarize outstanding problems and issues with protocols and studies. Determine if deficiencies were adequately addressed. Review statistical methodology of studies. Author all written communication to the sponsoring firm and integrate comments from other reviewers.
Education
Ph.D. – Physiology, University of Georgia – Athens, GA
M.S. – Physiology University of Maryland – College Park, MD
B.A. – Biology, University of Iowa – Iowa City, IA
A Partial List of Dr. Sharlin’s FDA Expert Witness Experience
- Explain how a generic drug company’s failure to meet FDA’s regulatory requirements for supplying a Medication Guide with a drug prescription supports a legal case for failure to warn.
- Analyze FDA’s drug safety database to show there was significant evidence of serious cardiovascular events among men taking hormone replacement therapy.
- Support a medical device company’s claim for monetary damages by showing how infringement of their patent solved a regulatory compliance problem of the defendant.
- For a hip implant, identify pre- and post-approval actions and inactions by the medical device manufacturer that were noncompliant with FDA’s regulatory requirements.
- Explain how a drug company’s manufacturing compliance failure negated their business loss insurance claim made after physical damage to their manufacturing facility.
- For a hip implant, describe the medical device manufacturer’s lack of FDA compliance in their efforts to obtain a 510(k) approval.
- Conduct an FDA compliance review of a medical device company’s actions after the installation of their implantable pulse generator (intended to relieve pain) caused harm.
- For a low risk Class I medical device, identify the company’s regulatory failures related to safety that resulted in a death.
- Write a report explaining why the Warning Section in the label for a generic version of sulindac was deficient.
- Write a report confirming a drug manufacturer’s processes for establishing the stability of a drug product met FDA’s compliance requirements.
- Review a clinical study intended to compare the effectiveness of two drugs. Show the study’s design was manipulated to mask the weakness of one drug over the other.
- Find weaknesses in a defense expert’s report on the adverse event reporting of fentanyl.
- Evaluate FDA compliance in reporting adverse event information about Actos.
- Deposed about the responsibilities of an Institutional Review Board in a wrongful firing case.
- Testify for the defendant in a fertility clinic damages case. Explain the process and predictability of FDA approval of a drug under development.
- Analyze the content of a drug IND submission to identify trade secret content.
- Identify FDA regulatory deficiencies in the adverse event reporting of a breast cancer drug and describe the resulting deficiencies in the drug’s label.
- Analyze the adverse event reporting for a drug that treats Attention Deficit Hyperactivity Disorder (ADHD) to determine if the drug’s label meets FDA regulatory requirements.
- For a schizophrenia drug, analyze the growing body of scientific literature about the drug and determine if the company’s updates to the drug label’s safety information were FDA compliant.
- Analyze FDA’s medical device safety database to show a company failed to report incidents about their medical device resulting in death.
- Explain how achieving drug development milestones triggered a disputed $100 million dollar payment in a merger agreement between two companies.